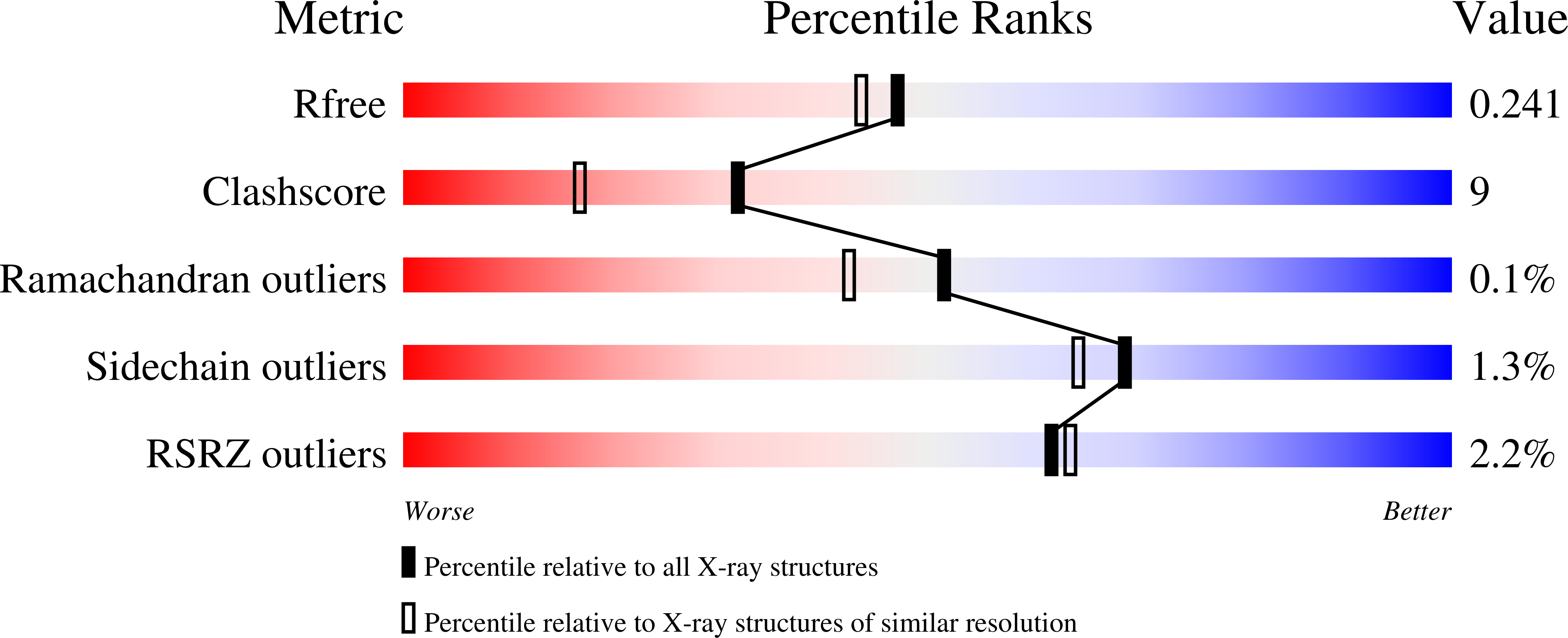
Double duty for a conserved glutamate in pyruvate decarboxylase: evidence of the participation in stereoelectronically controlled decarboxylation and in protonation of the nascent carbanion/enamine intermediate .
Meyer, D., Neumann, P., Parthier, C., Friedemann, R., Nemeria, N., Jordan, F., Tittmann, K.(2010) Biochemistry 49: 8197-8212
- PubMed: 20715795
- DOI: https://doi.org/10.1021/bi100828r
- Primary Citation of Related Structures:
3OE1 - PubMed Abstract:
Pyruvate decarboxylase (PDC) catalyzes the nonoxidative decarboxylation of pyruvate into acetaldehyde and carbon dioxide and requires thiamin diphosphate (ThDP) and a divalent cation as cofactors. Recent studies have permitted the assignment of functional roles of active site residues; however, the underlying reaction mechanisms of elementary steps have remained hypothetical. Here, a kinetic and thermodynamic single-step analysis in conjunction with X-ray crystallographic studies of PDC from Zymomonas mobilis implicates active site residue Glu473 (located on the re-face of the ThDP thiazolium nucleus) in facilitating both decarboxylation of 2-lactyl-ThDP and protonation of the 2-hydroxyethyl-ThDP carbanion/enamine intermediate. Variants carrying either an isofunctional (Glu473Asp) or isosteric (Glu473Gln) substitution exhibit a residual catalytic activity of less than 0.1% but accumulate different intermediates at the steady state. Whereas the predecarboxylation intermediate 2-lactyl-ThDP is accumulated in Glu473Asp because of a 3000-fold slower decarboxylation compared to that of the wild-type enzyme, Glu473Gln is not impaired in decarboxylation but generates a long-lived 2-hydroxyethyl-ThDP carbanion/enamine postdecarboxylation intermediate. CD spectroscopic analysis of the protonic and tautomeric equilibria of the cocatalytic aminopyrimidine part of ThDP indicates that an acidic residue is required at position 473 for proper substrate binding. Wild-type PDC and the Glu473Asp variant bind the substrate analogue acetylphosphinate with the same affinity, implying a similar stabilization of the predecarboxylation intermediate analogue on the enzyme, whereas Glu473Gln fails to bind the analogue. The X-ray crystallographic structure of 2-lactyl-ThDP trapped in the decarboxylation-deficient variant Glu473Asp reveals a common stereochemistry of the intermediate C2α stereocenter; however, the scissile C2α-C(carboxylate) bond deviates by ∼25-30° from the perpendicular "maximum overlap" orientation relative to the thiazolium ring plane as commonly observed in ThDP enzymes. Because a reactant-state stabilization of the predecarboxylation intermediate can be excluded to account for the slower decarboxylation, the data suggest a strong stereoelectronic effect for the transition state of decarboxylation as supported by additional DFT studies on models. To the best of our knowledge, this is a very rare example in which the magnitude of a stereoelectronic effect could be experimentally estimated for an enzymatic system. Given that variant Glu473Gln is not decarboxylation-deficient, electrostatic stress can be excluded as a driving force for decarboxylation. The apparent dual function of Glu473 further suggests that decarboxylation and protonation of the incipient carbanion are committed and presumably proceed in the same transition state.
Organizational Affiliation:
Albrecht-von-Haller-Institute for Plant Sciences and Göttingen Center for Molecular Biosciences, Georg-August-University Göttingen, Ernst-Caspari-Haus, Justus-von-Liebig-Weg 11, D-37077 Göttingen, Germany.