



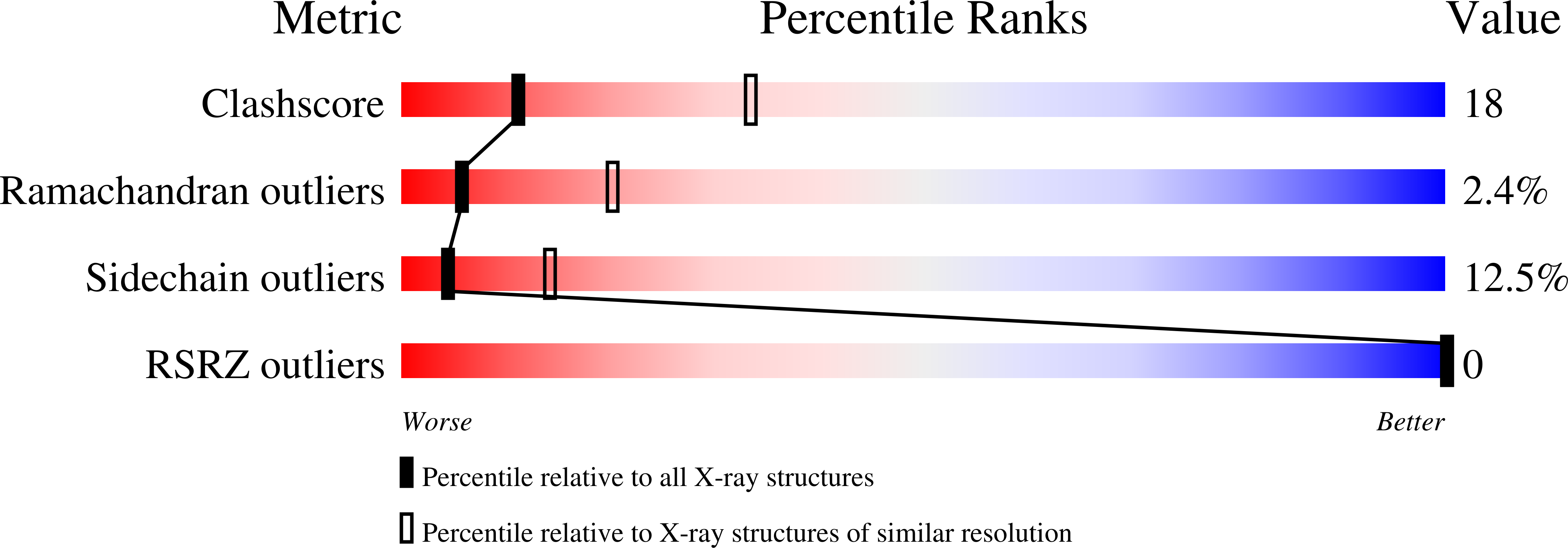
Glutathione reductase turned into trypanothione reductase: structural analysis of an engineered change in substrate specificity.
Stoll, V.S., Simpson, S.J., Krauth-Siegel, R.L., Walsh, C.T., Pai, E.F.(1997) Biochemistry 36: 6437-6447
- PubMed: 9174360
- DOI: https://doi.org/10.1021/bi963074p
- Primary Citation of Related Structures:
1GRT, 2GRT, 3GRT, 4GRT, 5GRT - PubMed Abstract:
Trypanosoma and Leishmania, pathogens responsible for diseases such as African sleeping sickness, Chagas' heart disease, or Oriental sore, are two of the very few genera that do not use the ubiquitous glutathione/glutathione reductase system to keep a stable cellular redox balance. Instead, they rely on trypanothione and trypanothione reductase to protect them from oxidative stress. Trypanothione reductase (TR) and the corresponding host enzyme, human red blood cell glutathione reductase (GR), belong to the same flavoprotein family. Despite their closely related three-dimensional structures and although their natural substrates share the common structural glutathione core, the two enzymes are mutually exclusive with respect to their disulfide substrates. This makes the parasite enzyme a potential target for antitrypanosomal drug design. While a large body of structural data on GR complexes is available, information on TR-ligand interactions is very limited. When the two amino acid changes Ala34Glu and Arg37Trp are introduced into human GR, the resulting mutant enzyme (GRTR) prefers trypanothione 700-fold over its original substrate, effectively converting a GR into a TR [Bradley, M., Bücheler, U. S., & Walsh, C. T. (1991) Biochemistry 30, 6124-6127]. The crystal structure of GRTR has been determined at 2.3 A resolution and refined to a crystallographic R factor of 20.9%. We have taken advantage of the ease with which ligand complexes can be produced in GR crystals, a property that extends to the isomorphous GRTR crystals, and have produced and analyzed crystals of GRTR complexes with glutathione, trypanothione, glutathionylspermidine and of a true catalytic intermediate, the mixed disulfide between trypanothione and the enzyme. The corresponding molecular structures have been characterized at resolutions between 2.3 and 2.8 A with R factors ranging from 17.1 to 19.7%. The results indicate that the Ala34Glu mutation causes steric hindrance leading to a large displacement of the side chain of Arg347. This movement combined with the change in charge introduced by the mutations modifies the binding cavity, forcing glutathione to adopt a nonproductive binding mode and permitting trypanothione and to a certain degree also the weak substrate glutathionylspermidine to assume a productive mode.
Organizational Affiliation:
Department of Biochemistry, University of Toronto, Ontario Cancer Institute, Canada.




