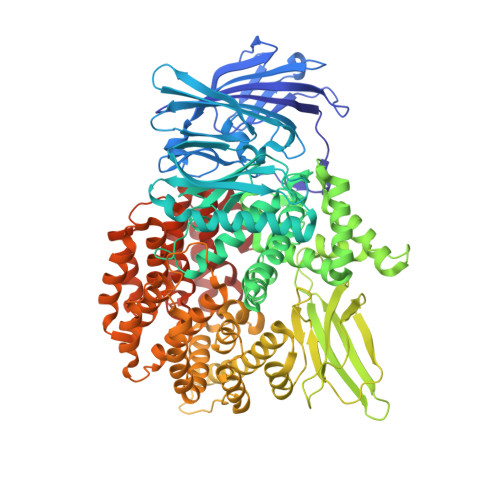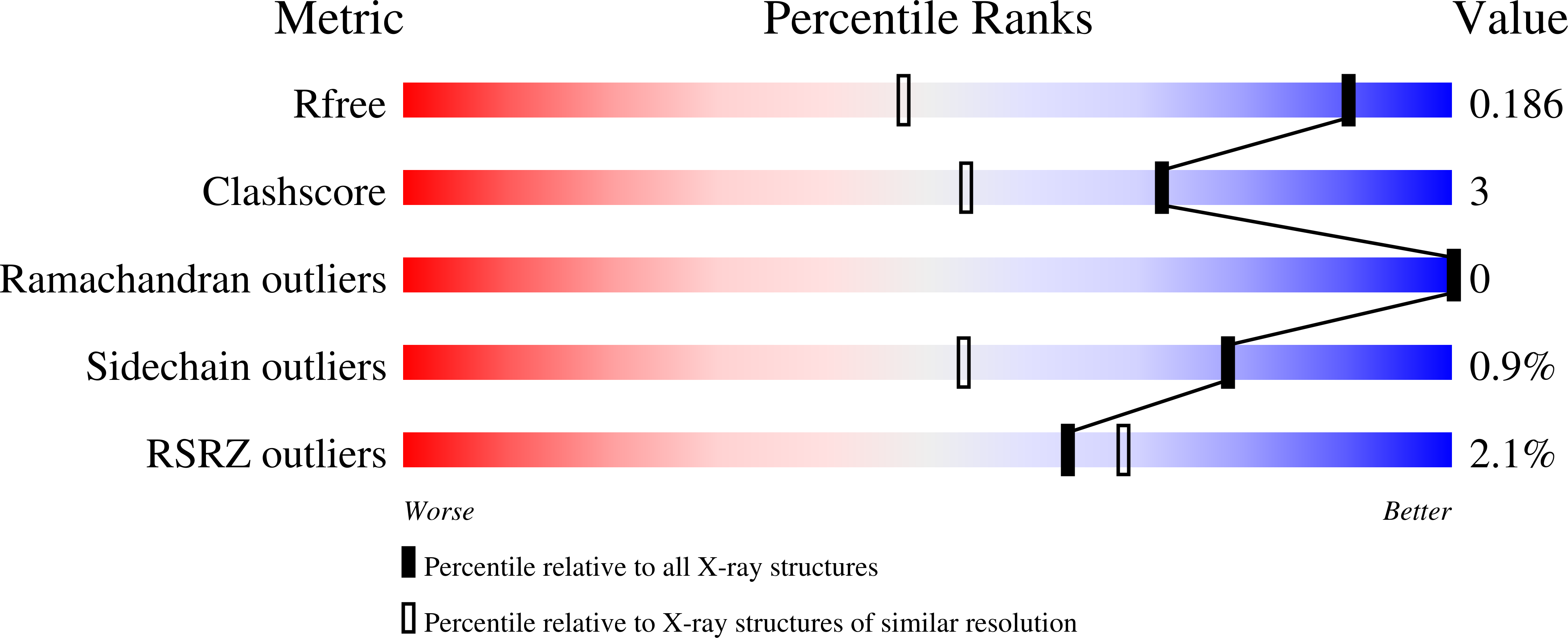
Hydroxamic Acid Inhibitors Provide Cross-Species Inhibition of Plasmodium M1 and M17 Aminopeptidases.
Vinh, N.B., Drinkwater, N., Malcolm, T.R., Kassiou, M., Lucantoni, L., Grin, P.M., Butler, G.S., Duffy, S., Overall, C.M., Avery, V.M., Scammells, P.J., McGowan, S.(2019) J Med Chem 62: 622-640
- PubMed: 30537832
- DOI: https://doi.org/10.1021/acs.jmedchem.8b01310
- Primary Citation of Related Structures:
6EA1, 6EA2, 6EAA, 6EAB, 6EE2, 6EE3, 6EE4, 6EE6, 6EED, 6EEE - PubMed Abstract:
There is an urgent clinical need for antimalarial compounds that target malaria caused by both Plasmodium falciparum and Plasmodium vivax. The M1 and M17 metalloexopeptidases play key roles in Plasmodium hemoglobin digestion and are validated drug targets. We used a multitarget strategy to rationally design inhibitors capable of potent inhibition of the M1 and M17 aminopeptidases from both P. falciparum ( Pf-M1 and Pf-M17) and P. vivax ( Pv-M1 and Pv-M17). The novel chemical series contains a hydroxamic acid zinc binding group to coordinate catalytic zinc ion/s, and a variety of hydrophobic groups to probe the S1' pockets of the four target enzymes. Structural characterization by cocrystallization showed that selected compounds utilize new and unexpected binding modes; most notably, compounds substituted with bulky hydrophobic substituents displace the Pf-M17 catalytic zinc ion. Excitingly, key compounds of the series potently inhibit all four molecular targets and show antimalarial activity comparable to current clinical candidates.
Organizational Affiliation:
Medicinal Chemistry, Monash Institute of Pharmaceutical Sciences , Monash University, Parkville , Melbourne , VIC 3052 , Australia.