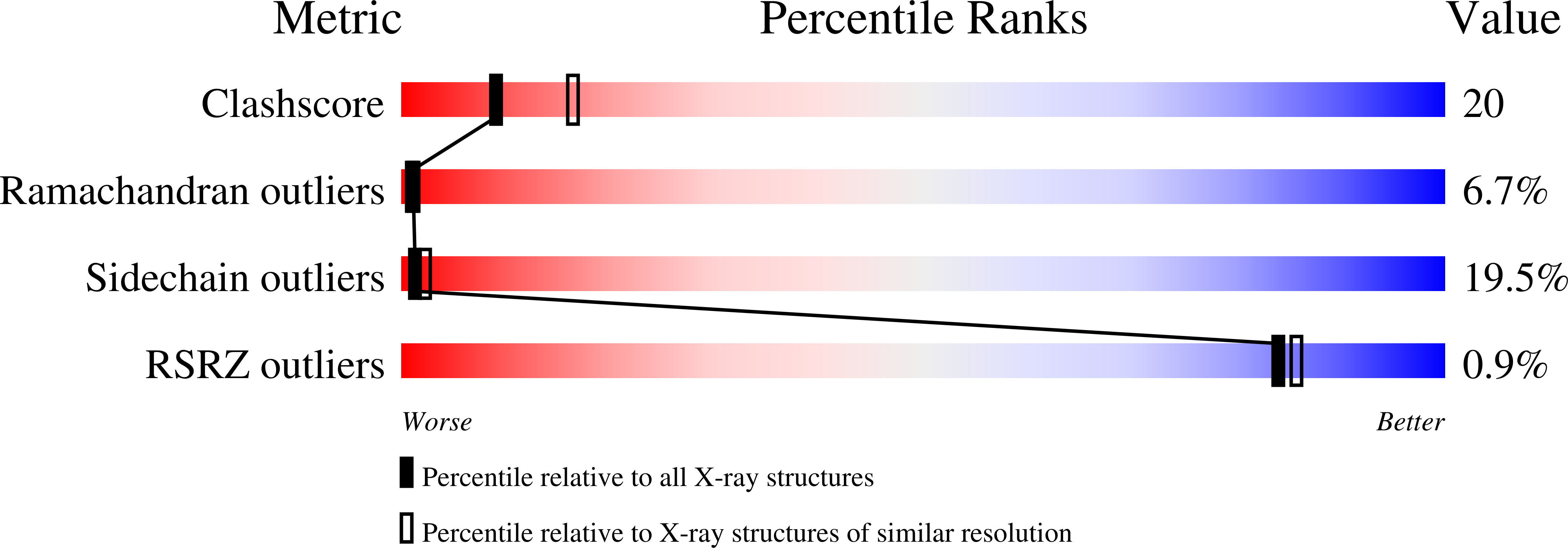
Refined structures of substrate-bound and phosphate-bound thymidylate synthase from Lactobacillus casei.
Finer-Moore, J., Fauman, E.B., Foster, P.G., Perry, K.M., Santi, D.V., Stroud, R.M.(1993) J Mol Biol 232: 1101-1116
- PubMed: 8371269
- DOI: https://doi.org/10.1006/jmbi.1993.1463
- Primary Citation of Related Structures:
1LCA, 1LCB, 1LCE, 1THY, 2TDM - PubMed Abstract:
Crystal structures of two crystal forms of the complex of Lactobacillus casei (TS) with its substrate dUMP have been solved and refined at 2.55 A resolution. The two crystal forms differ by approximately 5% in the c-axis length. The TS-dUMP complexes are symmetric dimers with dUMP bound equivalently in both active sites. dUMP is non-covalently bound in the same conformation as in ternary complexes of TS with dUMP and cofactor or cofactor analogs. The same hydrogen bonds are made between TS and substrate in the binary and ternary complexes. We have also determined the 2.36 A crystal structure of phosphate-bound L. casei TS. This structure has been refined to an R-factor of 19.3% with highly constrained geometry. Refinement has revealed the locations of all residues in the protein, including the disordered residues 90 to 119, which are part of an insert found only in the L. casei and Staphylococcus aureus transposon Tn4003 TS sequences. The 2.9 A multiple isomorphous replacement (MIR) structure of L. casei TS in a complex with its substrate dUMP has been refined to a crystallographic R-factor of 15.5%. Reducing agents were withheld from crystallization solutions during MIR structure determination to allow heavy-metal labeling of the cysteine residues. Therefore, the active-site cysteine residue in this structure is oxidized and the dUMP is found at half-occupancy in the active site. No significant conformational difference was found between the phosphate-bound and dUMP-bound structures. The TS-dUMP structures were better ordered than the phosphate-bound TS or the oxidized TS-dUMP, particularly Arg23, which is clearly hydrogen-bonded to the phosphate group of dUMP. A large and a small P6(1)22 crystal form are observed for both phosphate-bound and dUMP-bound L. casei TS. The small cell forms of the phosphate-bound and dUMP-bound enzyme are isomorphous, whereas the cell constants of the larger cell form change slightly when dUMP is bound (c = 240 A versus c = 243 A). For both liganded and unliganded enzyme, conversion from the small to the large crystal form sometimes occurs spontaneously, and the crystal packing changes at a single interface. Conversion may be the result of a small change in pH in the mother liquor surrounding the crystal. A single intermolecular contact between symmetry-related Asp287 residues is disrupted on going from the small to the large crystal form.
Organizational Affiliation:
Department of Biochemistry and Biophysics, University of California, San Francisco 94143-0448.