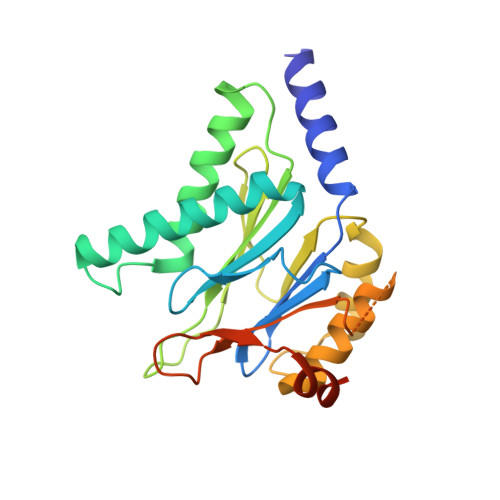
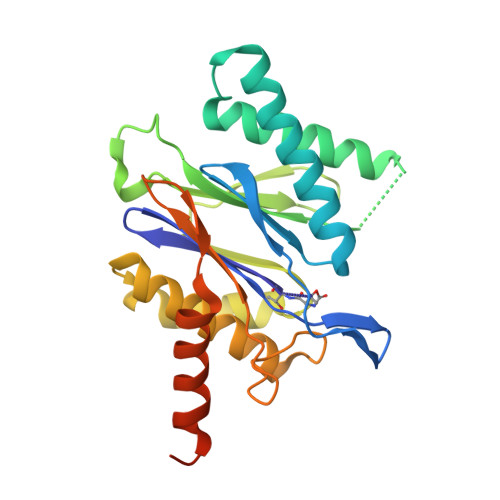
Inhibitors selective for mycobacterial versus human proteasomes.
Lin, G., Li, D., de Carvalho, L.P., Deng, H., Tao, H., Vogt, G., Wu, K., Schneider, J., Chidawanyika, T., Warren, J.D., Li, H., Nathan, C.(2009) Nature 461: 621-626
- PubMed: 19759536
- DOI: https://doi.org/10.1038/nature08357
- Primary Citation of Related Structures:
3H6F, 3H6I, 3HF9, 3HFA - PubMed Abstract:
Many anti-infectives inhibit the synthesis of bacterial proteins, but none selectively inhibits their degradation. Most anti-infectives kill replicating pathogens, but few preferentially kill pathogens that have been forced into a non-replicating state by conditions in the host. To explore these alternative approaches we sought selective inhibitors of the proteasome of Mycobacterium tuberculosis. Given that the proteasome structure is extensively conserved, it is not surprising that inhibitors of all chemical classes tested have blocked both eukaryotic and prokaryotic proteasomes, and no inhibitor has proved substantially more potent on proteasomes of pathogens than of their hosts. Here we show that certain oxathiazol-2-one compounds kill non-replicating M. tuberculosis and act as selective suicide-substrate inhibitors of the M. tuberculosis proteasome by cyclocarbonylating its active site threonine. Major conformational changes protect the inhibitor-enzyme intermediate from hydrolysis, allowing formation of an oxazolidin-2-one and preventing regeneration of active protease. Residues outside the active site whose hydrogen bonds stabilize the critical loop before and after it moves are extensively non-conserved. This may account for the ability of oxathiazol-2-one compounds to inhibit the mycobacterial proteasome potently and irreversibly while largely sparing the human homologue.
Organizational Affiliation:
Department of Microbiology and Immunology, Weill Cornell Medical College, New York, New York 10065, USA. [email protected]