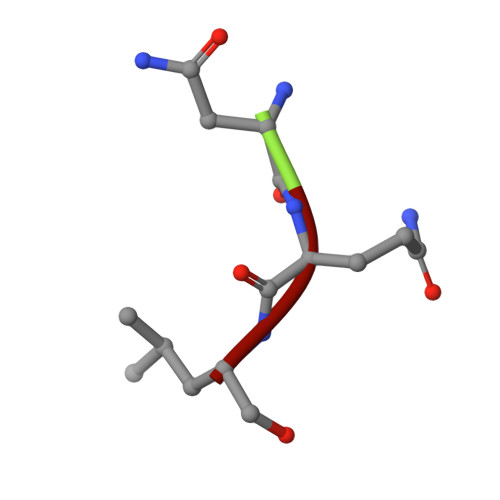
Fellutamide B is a potent inhibitor of the Mycobacterium tuberculosis proteasome.
Lin, G., Li, D., Chidawanyika, T., Nathan, C., Li, H.(2010) Arch Biochem Biophys 501: 214-220
- PubMed: 20558127
- DOI: https://doi.org/10.1016/j.abb.2010.06.009
- Primary Citation of Related Structures:
3KRD - PubMed Abstract:
Via high-throughput screening of a natural compound library, we have identified a lipopeptide aldehyde, fellutamide B (1), as the most potent inhibitor of the Mycobacterium tuberculosis (Mtb) proteasome tested to date. Kinetic studies reveal that 1 inhibits both Mtb and human proteasomes in a time-dependent manner under steady-state condition. Remarkably, 1 inhibits the Mtb proteasome in a single-step binding mechanism with K(i)=6.8 nM, whereas it inhibits the human proteasome beta5 active site following a two-step mechanism with K(i)=11.5 nM and K(i)(*)=0.93 nM. Co-crystallization of 1 bound to the Mtb proteasome revealed a structural basis for the tight binding of 1 to the active sites of the Mtb proteasome. The hemiacetal group of 1 in the Mtb proteasome takes the (R)-configuration, whereas in the yeast proteasome it takes the (S)-configuration, indicating that the pre-chiral CHO group of 1 binds to the active site Thr1 in a different orientation. Re-examination of the structure of the yeast proteasome in complex with 1 showed significant conformational changes at the substrate-binding cleft along the active site. These structural differences are consistent with the different kinetic mechanisms of 1 against Mtb and human proteasomes.
Organizational Affiliation:
Department of Microbiology and Immunology, Weill Medical College of Cornell University, 1300 York Ave., New York, NY 10065, United States. [email protected]