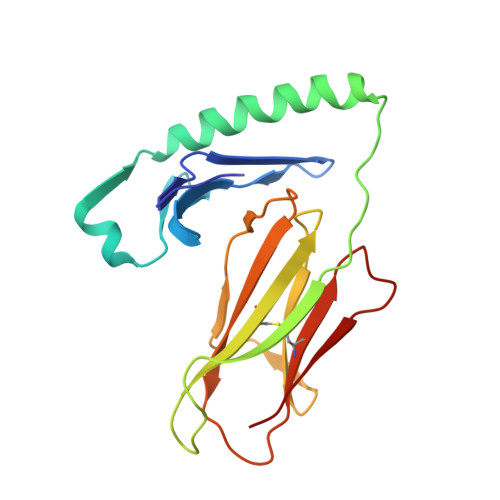
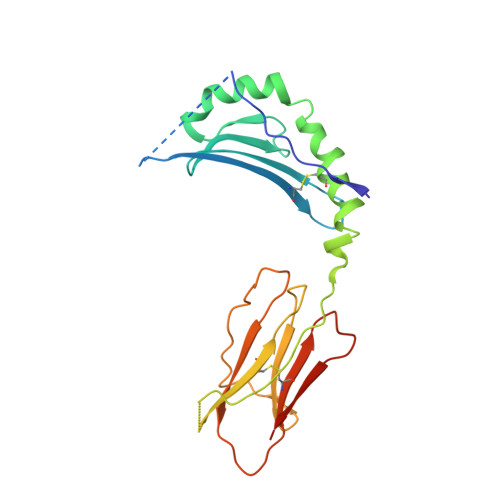
How C-terminal additions to insulin B-chain fragments create superagonists for T cells in mouse and human type 1 diabetes.
Wang, Y., Sosinowski, T., Novikov, A., Crawford, F., White, J., Jin, N., Liu, Z., Zou, J., Neau, D., Davidson, H.W., Nakayama, M., Kwok, W.W., Gapin, L., Marrack, P., Kappler, J.W., Dai, S.(2019) Sci Immunol 4
- PubMed: 30952805
- DOI: https://doi.org/10.1126/sciimmunol.aav7517
- Primary Citation of Related Structures:
6DFQ, 6DFS, 6DFV, 6DFW, 6DFX - PubMed Abstract:
In type 1 diabetes (T1D), proinsulin is a major autoantigen and the insulin B:9-23 peptide contains epitopes for CD4 + T cells in both mice and humans. This peptide requires carboxyl-terminal mutations for uniform binding in the proper position within the mouse IA g7 or human DQ8 major histocompatibility complex (MHC) class II (MHCII) peptide grooves and for strong CD4 + T cell stimulation. Here, we present crystal structures showing how these mutations control CD4 + T cell receptor (TCR) binding to these MHCII-peptide complexes. Our data reveal stricking similarities between mouse and human CD4 + TCRs in their interactions with these ligands. We also show how fusions between fragments of B:9-23 and of proinsulin C-peptide create chimeric peptides with activities as strong or stronger than the mutated insulin peptides. We propose transpeptidation in the lysosome as a mechanism that could accomplish these fusions in vivo, similar to the creation of fused peptide epitopes for MHCI presentation shown to occur by transpeptidation in the proteasome. Were this mechanism limited to the pancreas and absent in the thymus, it could provide an explanation for how diabetogenic T cells escape negative selection during development but find their modified target antigens in the pancreas to cause T1D.
Organizational Affiliation:
Department of Biomedical Research, National Jewish Health, Denver, CO 80206, USA.