X-ray Structures of the Proprotein Convertase Furin Bound with Substrate Analogue Inhibitors Reveal Substrate Specificity Determinants beyond the S4 Pocket.
Dahms, S.O., Hardes, K., Steinmetzer, T., Than, M.E.(2018) Biochemistry 57: 925-934
- PubMed: 29314830
- DOI: https://doi.org/10.1021/acs.biochem.7b01124
- Primary Citation of Related Structures:
6EQV, 6EQW, 6EQX - PubMed Abstract:
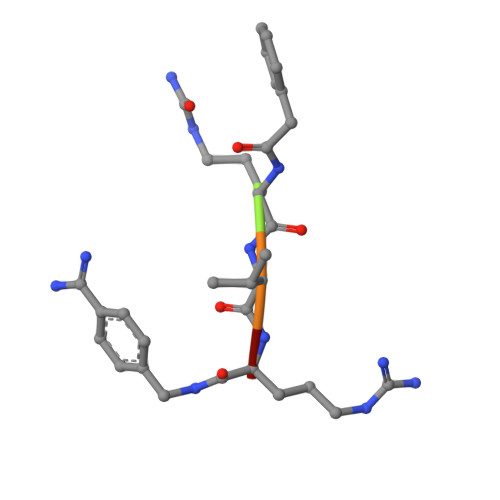
The proprotein convertase furin is a highly specific serine protease modifying and thereby activating proteins in the secretory pathway by proteolytic cleavage. Its substrates are involved in many diseases, including cancer and infections caused by bacteria and viruses. Understanding furin's substrate specificity is crucially important for the development of pharmacologically applicable inhibitors. Using protein X-ray crystallography, we investigated the extended substrate binding site of furin in complex with three peptide-derived inhibitors at up to 1.9 Å resolution. The structure of the protease bound with a hexapeptide inhibitor revealed molecular details of its S6 pocket, which remained completely unknown so far. The arginine residue at P6 induced an unexpected turnlike conformation of the inhibitor backbone, which is stabilized by intra- and intermolecular H-bonds. In addition, we confirmed the binding of arginine to the previously proposed S5 pocket (S5 1 ). An alternative S5 site (S5 2 ) could be utilized by shorter side chains as demonstrated for a 4-aminomethyl-phenylacetyl residue, which shows steric properties similar to those of a lysine side chain. Interestingly, we also observed binding of a peptide with citrulline at P4 substituting for the highly conserved arginine. The structural data might indicate an unusual protonation state of Asp264 maintaining the interaction with uncharged citrulline. The herein identified molecular interaction sites at P5 and P6 can be utilized to improve next-generation furin inhibitors. Our data will also help to predict furin substrates more precisely on the basis of the additional specificity determinants observed for P5 and P6.
Organizational Affiliation:
Department of Molecular Biology, University of Salzburg , Billrothstrasse 11, A-5020 Salzburg, Austria.