Molecular-dynamics simulation methods for macromolecular crystallography.
Wych, D.C., Aoto, P.C., Vu, L., Wolff, A.M., Mobley, D.L., Fraser, J.S., Taylor, S.S., Wall, M.E.(2023) Acta Crystallogr D Struct Biol 79: 50-65
- PubMed: 36601807
- DOI: https://doi.org/10.1107/S2059798322011871
- Primary Citation of Related Structures:
7UJX, 7V0G - PubMed Abstract:
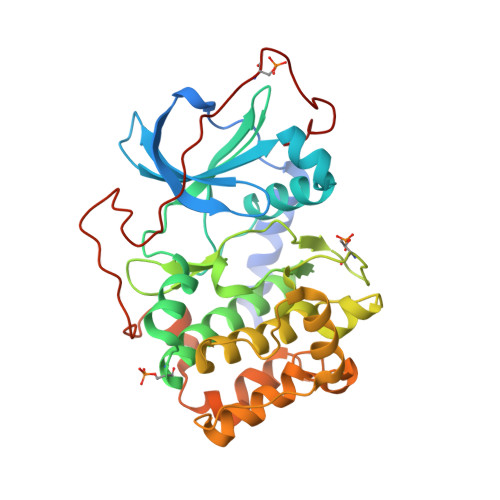
It is investigated whether molecular-dynamics (MD) simulations can be used to enhance macromolecular crystallography (MX) studies. Historically, protein crystal structures have been described using a single set of atomic coordinates. Because conformational variation is important for protein function, researchers now often build models that contain multiple structures. Methods for building such models can fail, however, in regions where the crystallographic density is difficult to interpret, for example at the protein-solvent interface. To address this limitation, a set of MD-MX methods that combine MD simulations of protein crystals with conventional modeling and refinement tools have been developed. In an application to a cyclic adenosine monophosphate-dependent protein kinase at room temperature, the procedure improved the interpretation of ambiguous density, yielding an alternative water model and a revised protein model including multiple conformations. The revised model provides mechanistic insights into the catalytic and regulatory interactions of the enzyme. The same methods may be used in other MX studies to seek mechanistic insights.
Organizational Affiliation:
Computer, Computational and Statistical Sciences Division, Los Alamos National Laboratory, Los Alamos, NM 87545, USA.